Using the Gen Bank Database
Updated 28/11/00
This Lab exercise
contains a Description of the needed source materials, a list of
objectives, a detailed description of each of the objectives and
the marking scheme
OBJECTIVES:
- Obtain Amino Acid
Sequences form the GenBank Database
- Install Clustal X on
your computer
- Align the amino acid sequences from the GenBank Database
using Clustal X.
- Analyse the polymorphism in the aligned sequences.
- Submit a report
Introduction
The mammalian immune system has innate and adaptive
components. The function of the adaptive immune system relies on
the ability of immune system to alter the amino acid sequences of
specific proteins in order to better recognise and defend against
specific pathogens. The sequences of these proteins is altered
over several generations in the case of Major Histocompatibility
Complex receptors , or over the course of a single infection in
the case of Immunoglobulins and T-Cell receptors . The presence
of many different forms of a specific protein within a population
of animals is call polymorphism. Modern molecular
biological and biochemical techniques allow researchers to study
these amino acid differences by sequencing genes or proteins from
different cells and individuals.and then comparing the sequences.
In this laboratory you will obtain several sequences from one of
the proteins mentioned above from an online database and analyse
their amino acid sequences in order to determine the degree of
polymorphism.
OBJECTIVE 1: Obtain amino acid Sequences
from the GenBank Database
READ THIS ENTIRE OBJECTIVE BEFORE YOU START !!!
In this objective you will learn to access the
National Center for Biotechnology Information website and use
their "Entrez" search engine to find and retrieve
protein sequences that will be used later in the exercise
To achieve this objective you will
- Access the website
- Become familar with the website and its search
engine
- Use a keyword search to obtain a list of files
with the appropriate protein sequences
- Open a text file in Notepad or Wordpad to store
the retrieved sequences
- Open a file from the NCBI database and find the
amino acid sequence
- Copy the amino acid sequence
- Paste the sequence into the Notepad text file
- Repeat the open, copy and
paste operation for 10 molecules
- Save the Notepad text file with the 10 different
protein sequences
- Close the NCBI database
- Format the text file to be used by the Clustal X
program
The National Center for
Biotechnology Information (NCBI) is an international repository
of data on the sequence of genes and proteins. Researchers from
around the world deposit their information here so that it can be
used by the scientific community. The data are stored in several
different databases; the "proteins" databases contain
just the amino acid sequences while the "Nucleotides"
databases contain both the nucleotide sequences and the deduced
amino acid sequences.You can use either of these databases to
retrieve the sequences that you will need for this exercise.
Data on millions of sequences are
stored in separate files each with a unique identification code. You
can use key-words to find specific sequences deposited in the
databases.
The NCBI is
constantly updated and modified as they find faster ways to
retreive the information. Do not be surprised if the screen
layout changes frequently as improvements ae brought online.
As you browse the
NCBI website look for answers to the following questions
- What is the
function of the NCBI?
- What
organizations support the NCBI?
- What kind of
information is stored in the NCBI?
- What is
GenBank?
- Who are the
members of the International Nucleotide sequence Database
collaboration?
- How many
species or represented in the database?
- How fast is
the database expanding?
RETRIEVING AMINO ACID SEQUENCES
Before you start your search make certain that you have space
on your computer to store the info that you retrieve. While you
can store the info on a hard drive, a 3 1/2" floppy disk
will be more than enough. As you will have to submit the data on
a floppy disk anyway, why not save it directly to floppy disk?
Also, before you start your search make certain that you have
opened a text file for future storage. Open a new text file in
NotePad or Wordpad
Use the NCBI Entrez search engine to obtain 10 amino
acid sequences of a specific protein from a single species and
cut and paste these sequences in the opened text file as
described below
You want to find specific sequences of proteins that are
immunologically important eg, MHC, T cell Receptors, Kappa chain,
Lambda chains or any of the heavy chains of immunoglobulins. Use
only sequences from a single isotype, for example IgM or IgG, and
from only the heavy or the light chain. The Entrez search engine
lets you use key words to find these
Examples of Key word searches
- "homo sapiens, MHC II" produces a list of MHCII
sequences
- "homo sapiens, MHC I" will produces a list of
MHCII sequences
- "homo sapiens, IgM, variable" will produce a
list of IgM variable domains.
- "homo sapiens, IgG, variable" will produce a
list of IgG variable domains
- "homo sapiens, Kappa, variable" will produce a
list of kappa chain variable domains
- "homo sapiens, Lambda, variable" will produce a
list of Lambda chain variable domains
After you have completed your search you will have a list of
files with the appropriate sequences
SAVING THE SEQUENCE DATA
- Select a file from the list that has been created by the
key word search
- Click on the GenPept ot GenBank report link to see the
full entry for a specific sequence. The amino acid
sequence (in one letter code) will be near the bottom.
- Copy this sequence by highlighting the sequence and using
the copy command (CTRL C is faster)
- Go to the text file that you opened in Notepad or Wordpad
and use the paste command (CTRL V is faster)
- Move on to another sequence and paste it in the same text
file until you have 10
- TIP try an pick sequences at are about the same size
- Save the text file
FORMATTNG THE TEXT FILE TO BE USED BY CLUSTAL X
The text file containing your sequences should be formatted to
meet the following:
CLUSTAL FORMAT RULES
- The font must be Courier, 10 point
- The >P1;
tells Clustal X that the following is a sequence for
alignment
- The words following on the same line after >P1;
indicate the sequence name. Use any name that you choose
but do not repeat the name. Use the Underscore reather
than the space
- Leave a blank line after the title line,
- The sequence must have amino acid letter codes only ( no
numbers)
- The sequence must end with a star (*).
- Save the file as "text only"
I usually call the file something that will indicate what I am
aligning. The example below would be "stvi.txt" .
EXAMPLE OF A CLUSTAL FORMATTED TEXT FILE
>P1;Stvi-10-8
HSLKFYCTASSGVPNFPEFVAVGLVDDVEISHYDSNTKREEPKQDWMSRVTEDDPQFWQRETEISEGHQQTFKANIEVVRQRLNQT*
>P1;Auha-706
HSLKYFYTASSQVPNFPEFVTVGMVDDVQVDYYDSDTEKAEPKQDWIARNTDQQYWERNTDIYRGSQQSFKANIEIVKQRFNQT*
>P1;Auha-705
HSLKFFCTETPGVQSIPEFVAVAFVDEVQIGDFNNVRGAEPKKDWIKFFADHPEHLEWYSSISKQSHQVFKANIETFRQRLNQT*
>P1;Auha-517
HSLKYFFTGSSQVPNFPEFVVVGMVDDVQVVHYDSDTEKAGPKQDWFARNTDQQYWESQTGNLLGSQQTFKANIETAKQRFNQT*
>P1;Cyca-UAA
HTLQYFYTATSGIENFPEFMTAGVVDGQQIDYYDSIIRKAVQKAEWISGAVDPDYWNRNTQIYAGNEPSFKENINIVKSRFNQT*
>P1;Furu-UAA
HTLKYFYTASSGVPNFPEFVIVGMVDDVQMVRYDSNTRRMQPKQEWMKEFTADDPQYLDTQSQNAFGAQQIYKANIEIAKERFNQT*
>P1;Sasa-UAA
HALKYFYTASSEVPNFPEFVVVGVVDGVQMVHYDSNSQRAVPKQDWVNKAADPQYWERNTGIFKGSQQTFKANIDIAKQRFNQS*
>P1;Plam-sp005
HSLKNFHTASSQVTNIPEFVFVGFIDDVQTEYYDSNTKKSEPKQDWMLKATDAKYWERQTGILRAYQRVFKANMEIAKERFNQT*
>P1;Stvi-10-9
HSLKFYFTAFFGVPNFPEFVSVGLVDDIEISRYDSNTKREEPTQDWMSRVPQEDPPFWPTETEISDGDQQTFQAHIEVVKQRLNQT*
>P1;Stvi-10-10
HSLKIFCTACSGVPNFPEYVSVGLVDGVQMIRYDSNTKRQEPKQDWMSRVTEDDPQYWQRNTEIAEGNQQVYKAGIEILKQRLNQT*
Submit a copy of your file with your report.
OBJECTIVE 2 Install Clustal X on your
Computer
DownLoad Clustal X
A copy of Clustal X is stored in
Immunology>>Resources
>>Software Downloads>> Clustal X under the file
name clustalx1_8msw.exe This is a compressed
document that has several files. This program will not work until
it has been downloaded and installed
To Download this file, goto the appropriate section on the
website, Double click on the filename, tell the computer where
you want to store the file on your hard drive. Remember where you
have stored it!
Install Clustal X
To install Clustal X, find the file clustalx1_8msw.exe
that is now stored on your computer. Double click on the
file name and follow the install instructions You will have to
tell the computer where you want the program ClustalX installed.
You could create a subdirectory called Clustal in the Program
Files directoryand install CLUSTALX in the subdirectory.An
executable file (clustalx.exe) which will run
under MS Windows (32 bit). The directory containing the
executable (plus the files named *.par,and clustalx.hlp) should
be added to your path defined in the autoexec.batfile.
Browse through the menu options and the help files of ClustalX
to learn some of its features
OBJECTIVE 3 Align the sequences using
Clustal X
download the file called Clustal_Example.txt
an use it as a test example .
Start the program Clustal X (clustalx.exe).
Select Multiple Alignment Mode
Select file
select load sequences
Find and select the file Clustal_Example.txt
If ClustalX recognises you file, it will list the sequences and
the number of amino acids in each one. (If it doesn’t do
this, check that your file is saved as text only).
The path and the filename should appear at the bottom
of the screen
A list of the alignment sequences will appear in the
left hand frame
The sequences will appear as multi coloured letter
codes on the right hand frame
Select the aligment option from the menu
Selct the Do Complete Alignment option
You will be asked for names for the alignment and dendrogram
files. Just use the default names, which will be the same as your
text file, with either an .aln (eg stvi.aln) or .dnd (eg
stvi.dnd) extension. The alignment will appear on the screen.
Close the clustal window, then using a wordprocessor such as
Microsoft Wordpad, Microsoft Word or Wordperfect open up the .aln
file (eg stvi.aln). Your alignment should be there and it should
look like the example below (note the example below is comparing
MHC class I sequences from several different fish species - your
sequences should be much more similar!). Deletions within the
sequence will be indicated by dashes. The bottom line of the
alignment will contain several symbols. Stars (*) indicate amino
acids which are identical, colons (:) indicate amino acid changes
which are conservative (hydrophobic for hydrophobic or acidic for
acidic), while periods (.) indicate changes which may be
conservative. If the rows do not line up, change the font to
Courier, 10 point.
CLUSTAL X (1.8) multiple
sequence alignment
Stvi-10-8
HSLKFYCTASSGVPNFPEFVAVGLVDDVEISHYDSNTKREEPKQDWMSRVTEDDPQFWQR
Stvi-10-9
HSLKFYFTAFFGVPNFPEFVSVGLVDDIEISRYDSNTKREEPTQDWMSRVPQEDPPFWPT
Stvi-10-10
HSLKIFCTACSGVPNFPEYVSVGLVDGVQMIRYDSNTKRQEPKQDWMSRVTEDDPQYWQR
Cyca-UAA
HTLQYFYTATSGIENFPEFMTAGVVDGQQIDYYDSIIRKAVQKAEWISG--AVDPDYWNR
Sasa-UAA
HALKYFYTASSEVPNFPEFVVVGVVDGVQMVHYDSNSQRAVPKQDWVNK--AADPQYWER
Auha-706
HSLKYFYTASSQVPNFPEFVTVGMVDDVQVDYYDSDTEKAEPKQDWIAR--NTDQQYWER
Auha-517
HSLKYFFTGSSQVPNFPEFVVVGMVDDVQVVHYDSDTEKAGPKQDWFAR--NTDQQYWES
Plam-sp005
HSLKNFHTASSQVTNIPEFVFVGFIDDVQTEYYDSNTKKSEPKQDWMLK--ATDAKYWER
Furu-UAA
HTLKYFYTASSGVPNFPEFVIVGMVDDVQMVRYDSNTRRMQPKQEWMKEFTADDPQYLDT
Auha-705
HSLKFFCTETPGVQSIPEFVAVAFVDEVQIGDFN-NVRGAEPKKDWIKFF-ADHPEHLEW
*:*: : * : .:**:: ...:* : ::
. . :*. . .
Stvi-10-8
ETEISEGHQQTFKANIEVVRQRLNQT
Stvi-10-9 ETEISDGDQQTFQAHIEVVKQRLNQT
Stvi-10-10 NTEIAEGNQQVYKAGIEILKQRLNQT
Cyca-UAA NTQIYAGNEPSFKENINIVKSRFNQT
Sasa-UAA NTGIFKGSQQTFKANIDIAKQRFNQS
Auha-706 NTDIYRGSQQSFKANIEIVKQRFNQT
Auha-517 QTGNLLGSQQTFKANIETAKQRFNQT
Plam-sp005 QTGILRAYQRVFKANMEIAKERFNQT
Furu-UAA QSQNAFGAQQIYKANIEIAKERFNQT
Auha-705 YSSISKQSHQVFKANIETFRQRLNQT
: . :: :: :.*:**:
OBJECTIVE 4: Analyse the polymorphism in the
aligned sequences
Researchers who study polymorphic molecules use a special
method to present and interpret their results, called a Wu-Kabat
variability plot. This is a graph which indicates the position
along the amino acid chain on the X axis and the variability
index along the Y axis. The variability index is calculated as
follows:
Number of different amino acids at a given
position along the amino acid chain
Frequency of the most common amino acid present
at a given position along the amino acid chain
For example in the alignment above position 1 is histidine (H)
in all ten sequences so the variability index is calculated as
follows:
Number of different amino acids = 1
Frequency of the most common =10 out of 10 = 10/10 = 1
Variability index = 1/1 = 1
In the alignment above position 2 is variable and the
variability index is calculated as follows:
Number of different amino acids (S, T, A) = 3
Frequency of the most common (S)= 7 out of 10 = 7/10 = 0.7
Variability index = 3/0.7 = 4.29
Position 21, which follows the sequence PEFV, is variable and
the variability index is calculated as follows:
Number of different amino acids (A,S,T,V,F,I)= 6
Frequency of the most common (A or S or T or V) =2 out of 10 =
2/10 = 0.2
Variability index = 6/0.2 = 30
In a Wu-Kabat plot these values are plotted against their
position in the polypeptide chain. An example of such a plot is
given below:
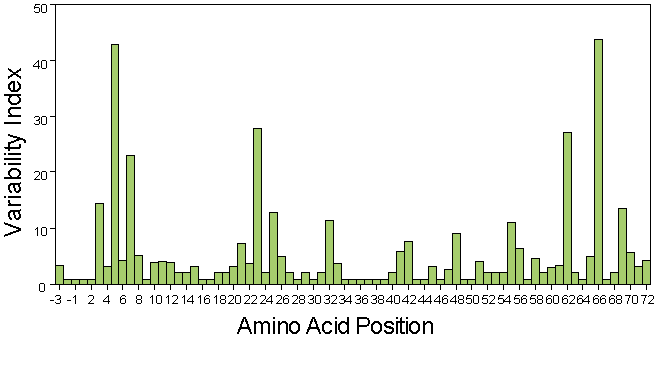
OBJECTIVE 5: Submit a Report
Construct a Wu-Kabat plot for the alignment you have produced.
Submit a report indicating the type of sequence you analysed
(MHC, immunoglobulin, T- Cell receptor) and the names of the
sequences you used. Include the your .pir file, your .aln file
and your Wu-Kabat plot. You can hand in all of these items in
electronic format on a 3.5 inch computer disk if you wish.
Appendix: The one
letter amino acid code
- G - Glycine (Gly)
- P - Proline (Pro)
- A - Alanine (Ala)
- V - Valine (Val)
- L - Leucine (Leu)
- I - Isoleucine (Ile)
- M - Methionine (Met)
- C - Cysteine (Cys)
- F - Phenylalanine (Phe)
- Y - Tyrosine (Tyr)
- W - Tryptophan (Trp)
- H - Histidine (His)
- K - Lysine (Lys)
- R - Arginine (Arg)
- Q - Glutamine (Gln)
- N - Asparagine (Asn)
- E - Glutamic Acid (Glu)
- D - Aspartic Acid (Asp)
- S - Serine (Ser)
- T - Threonine (Thr)
CLUSTAL REFERENCES
Details of algorithms, implementation and useful tips on usage of
Clustal
programs can be found in the following publications:
Jeanmougin,F., Thompson,J.D., Gouy,M., Higgins,D.G. and
Gibson,T.J. (1998)
Multiple sequence alignment with Clustal X. Trends Biochem Sci,
23, 403-5.
Thompson,J.D., Gibson,T.J., Plewniak,F., Jeanmougin,F. and
Higgins,D.G. (1997)
The ClustalX windows interface: flexible strategies for multiple
sequence
alignment aided by quality analysis tools. Nucleic Acids
Research, 24:4876-4882.
Higgins, D. G., Thompson, J. D. and Gibson, T. J. (1996) Using
CLUSTAL for
multiple sequence alignments. Methods Enzymol., 266, 383-402.
Thompson, J.D., Higgins, D.G. and Gibson, T.J. (1994) CLUSTAL W:
improving the
sensitivity of progressive multiple sequence alignment through
sequence
weighting, positions-specific gap penalties and weight matrix
choice. Nucleic
Acids Research, 22:4673-4680.
Higgins,D.G., Bleasby,A.J. and Fuchs,R. (1992) CLUSTAL V:
improved software for
multiple sequence alignment. CABIOS 8,189-191.
Higgins,D.G. and Sharp,P.M. (1989) Fast and sensitive multiple
sequence
alignments on a microcomputer. CABIOS 5,151-153.